
Summary
Large-vessel vasculitis (LVV), including Takayasu arteritis (TA), is a rare inflammatory disorder that can present with a wide range of clinical manifestations, often making diagnosis challenging. We report the case of a 56-year-old woman who presented with a one-month history of prolonged fever, progressive dyspnea, loss of appetite, and weight loss. Initial examination revealed an early diastolic murmur, raising a strong clinical suspicion of infective endocarditis. Further clinical assessment revealed moderate to severe aortic regurgitation, unequal blood pressure measurements between the two arms, and audible carotid and femoral bruits, suggesting large-vessel involvement. Blood cultures were repeatedly sterile, and transthoracic echocardiography showed no evidence of valvular vegetations, making infective endocarditis unlikely. Laboratory investigations revealed markedly elevated inflammatory markers, indicating an active systemic inflammatory process.
In view of the unequal pulses, inter-arm blood pressure discrepancy, and the presence of pericardial effusion on echocardiography, contrast-enhanced computed tomography of the chest was performed to exclude aortic dissection. Subsequent CT aortography demonstrated mural thickening and luminal narrowing of the left common carotid and left subclavian arteries, findings consistent with large-vessel vasculitis. Autoimmune serologies were positive for high-titre antinuclear antibodies, anti-SSA, and anti-Ro52, leading to a diagnosis of connective tissue disease-associated large-vessel vasculitis, specifically Takayasu arteritis with concurrent Sjogren’s syndrome. This case highlights the diagnostic complexity of TA, particularly when associated with another autoimmune disease and presenting with uncommon cardiovascular manifestations such as significant aortic regurgitation.
Background
Large-vessel vasculitis (LVV) is a group of chronic inflammatory disorders primarily affecting the aorta and its major branches. The two main forms are Takayasu arteritis (TA) and Giant Cell Arteritis (GCA). TA typically affects young women under 40 years of age, often in Asian populations, and can lead to arterial stenosis, occlusion, aneurysm formation, and aortic valve involvement. Patients commonly present with constitutional symptoms such as fever, weight loss, fatigue, and arthralgia during the early inflammatory phase. These are often followed by vascular manifestations including diminished pulses, unequal blood pressure, arterial bruits, hypertension, and ischemic symptoms. Cardiac complications, particularly aortic regurgitation, can occur due to inflammation and dilation of the ascending aorta and aortic root. Diagnosis relies on clinical findings, elevated inflammatory markers, advanced vascular imaging (e.g., CT or MR angiography), and the exclusion of infectious causes and aortic dissection. The presence of autoimmune markers may indicate an association with underlying connective tissue diseases. Early recognition of the disease and prompt initiation of immunosuppressive therapy are essential to prevent disease progression and irreversible vascular damage.
Case Presentation
A 56-year-old female presented with a one-month history of weight loss, fever and five days of progressive shortness of breath. There was no prior history of connective tissue disease or other significant medical illnesses. Initial examination revealed a temperature of 102°F (38.9°C) and an early diastolic murmur (EDM). The initial provisional diagnosis considered was aortic regurgitation secondary to infective endocarditis, given the fever, new murmur, and symptoms of heart failure.
However, a detailed assessment for classical peripheral signs of chronic severe aortic regurgitation yielded atypical findings: pulse volume was moderate, with no collapsing pulse, making classical chronic valvular AR less convincing. Further examination uncovered several atypical vascular signs, including forceful carotid pulsation, unequal pulses and blood pressure in both upper limbs, and bilateral carotid and femoral bruits. These findings strongly suggested pathology involving the aortic root or large vessels rather than an isolated valvular issue. Echocardiography demonstrated moderate to severe aortic regurgitation, a pericardial effusion of 1.2 cm without tamponade and a preserved ejection fraction of 65%. Crucially, no vegetations or spontaneous echo contrast were observed, making infective endocarditis less likely.
The combination of unequal pulses and blood pressure, vascular bruits, and pericardial effusion shifted the focus towards an aortic root pathology. Acute aortic dissection was a critical differential diagnosis due to the AR, pulse asymmetry, and blood pressure discrepancies and pericardia effusion on Echocardiogram. A contrast-enhanced computed tomography (CECT) of the chest was performed, which excluded aortic dissection and aneurysmal dilatation. However, it demonstrated cardiomegaly with pericardial effusion, mediastinal lymphadenopathy, and active-on-chronic chest infection. The constellation of pyrexia of unknown origin (PUO), multiple vascular bruits, unequal pulses and blood pressure, aortic regurgitation likely originating from root disease, and high inflammatory markers (ESR = 70 mm/hr, CRP = 160 mg/L, reactive thrombocytosis) raised strong suspicion for large-vessel vasculitis, particularly Takayasu arteritis. The patient was initiated on intravenous methylprednisolone 40 mg every 12 hours, which led to normalization of her temperature within 24 hours and throughout her hospital stay.
Investigations and Diagnostic Workup
Following the strong clinical suspicion, a CT aortogram was performed, which demonstrated narrowing and mural thickening of the left common carotid artery and left subclavian artery, without calcification, consistent with vasculitis. Additional findings included pericardial effusion, cardiomegaly, right middle lobe consolidation, and para-aortic lymphadenopathy. A PET scan further supported the diagnosis of large vessel vasculitis.
Further investigations into autoimmune markers revealed a positive antinuclear antibody (ANA) at a titre of 1:1280 (cytoplasmic pattern), positive extractable nuclear antigen (ENA) for SSA, and strongly positive Ro52, suggesting an underlying autoimmune inflammatory process. Based on these findings, the patient was diagnosed with connective tissue disease-associated large-vessel vasculitis, most consistent with Takayasu arteritis with concurrent Sjögren’s syndrome. Although the patient did not report any sicca symptoms, further evaluation for Sjögren’s syndrome was undertaken in view of the strongly positive autoimmune serology. Subsequent Schirmer’s testing was positive, supporting the diagnosis.
The patient was treated with intravenous methylprednisolone at a high dose for five consecutive days, followed by oral prednisolone at 1 mg/kg/day. A marked clinical response was observed, with fever and constitutional symptoms resolving after the second dose of intravenous methylprednisolone. She remained afebrile thereafter, and her general well-being returned to baseline prior to discharge. Serial laboratory assessments demonstrated normalization of inflammatory markers following corticosteroid therapy and intravenous antibiotic treatment.
Before initiating long-term immunosuppressive therapy, further evaluation was undertaken because of pulmonary consolidation identified on CT imaging and increased uptake on PET-CT. Following multidisciplinary discussion with the respiratory team, the possibility of underlying pulmonary tuberculosis could not be confidently excluded despite the absence of respiratory symptoms, negative GeneXpert and normalization of inflammatory markers. Consequently, anti-tuberculous therapy was commenced before the introduction of long-term immunosuppressive treatment.
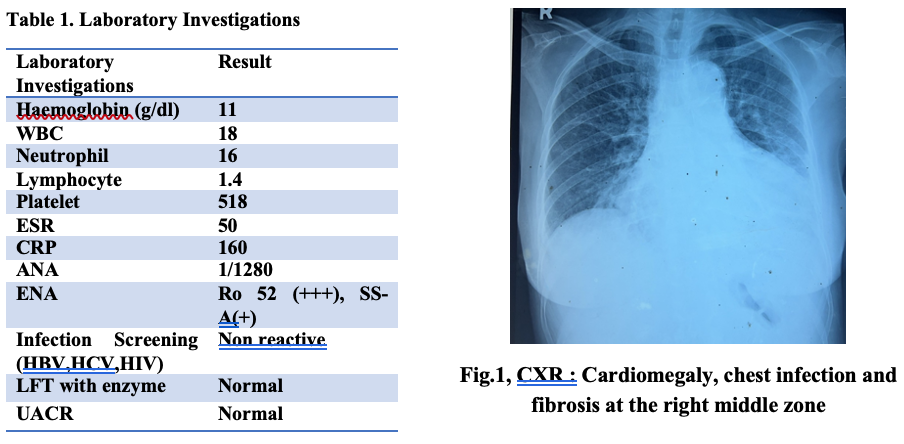
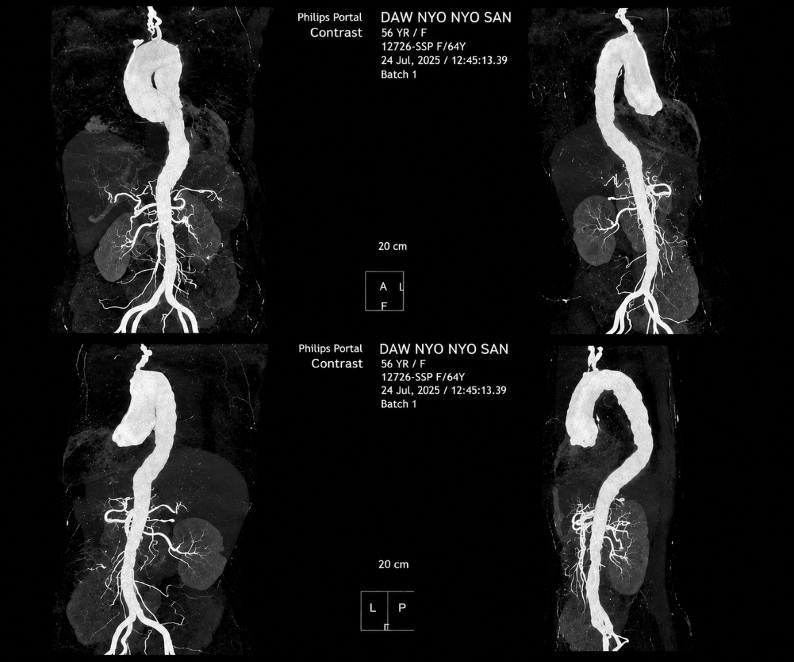
Fig. 2, CT aortogram: narrowing and mural thickening without calcification in left common carotid and left subclavian arteries
Discussion
This case presents a rare and diagnostically challenging scenario of Takayasu arteritis (TA) coexisting with Sjogren’s syndrome (SjS), manifesting primarily with severe aortic regurgitation (AR) and constitutional symptoms. Aortic regurgitation is most commonly attributed to primary valvular pathology; however, diseases affecting the aortic root and ascending aorta, such as large-vessel vasculitis, can also lead to AR through annular dilatation or inflammatory destruction of the aortic wall 2. The initial presentation with prolonged fever, a new early diastolic murmur, and heart failure symptoms typically prompts evaluation for infective endocarditis (IE). However, the absence of classical peripheral signs of chronic severe AR (e.g., collapsing pulse, wide pulse pressure) and sterile echocardiographic findings without vegetations made IE less probable. The presence of marked asymmetry in upper limb pulses and blood pressure, along with carotid and femoral bruits redirecting the diagnostic pathway towards large-vessel involvement. These clinical signs are characteristic of TA and reflect stenotic lesions in the aortic arch branches. Acute aortic dissection was also a significant differential diagnosis given the AR, pulse asymmetry, and blood pressure discrepancies. Timely cross-sectional imaging, specifically a CECT chest, effectively excluded dissection and aneurysmal disease, while a subsequent CT aortogram confirmed inflammatory mural thickening and luminal narrowing consistent with large-vessel vasculitis. The finding of pericardial effusion further supported a systemic inflammatory process, reducing the likelihood of isolated valvular pathology.
Although the patient was diagnosed with Takayasu arteritis, the age at presentation (56 years) was unusual, as this disease typically occurs in individuals younger than 50 years. However, the pattern of extensive aortic branch involvement, constitutional symptoms, and imaging findings were highly suggestive of TA, which classically affects younger women but can have atypical late-onset presentations. The distinction between TA and GCA can be challenging due to overlapping clinical and radiological features.
Cardiovascular involvement, particularly AR, is a major determinant of morbidity and mortality in TA, occurring in 20%-24% of patients of TA 2. The aortic regurgitation observed in Takayasu arteritis is primarily attributable to aortic root and annular dilatation secondary to chronic aortic inflammation, resulting in failure of valve leaflet coaptation. Direct inflammatory involvement of the valve leaflets may also contribute in some patients. 10,11,12. The markedly elevated inflammatory markers (ESR, CRP) and reactive thrombocytosis further corroborated active vascular inflammation.
An unusual aspect of this case was the coexistence of positive autoimmune serology, including high-titre ANA, anti-SSA, and strongly positive anti-Ro52. While not diagnostic for LVV, these antibodies indicate underlying immune dysregulation and are commonly associated with Sjögren’s syndrome 5. The concurrence of SjS and TA is exceptionally rare, with limited documented cases 1. This association suggests potential shared immune-mediated pathogenic mechanisms and highlights the importance of considering LVV in SjS patients who develop new systemic or vascular symptoms 5. Our patient’s presentation was atypical for SjS, with prominent vasculitis symptoms and no initial sicca symptoms, which are usually the hallmark of SjS. Furthermore, vasculitis associated with Sjögren’s syndrome (SjS) predominantly affects small- and medium-sized vessels, whereas large-vessel involvement is an uncommon extraglandular manifestation 4. Anti-Ro52 antibodies have been associated with activation of interferon-mediated immune pathways, and emerging evidence suggests that interferon signaling plays an important role in the pathogenesis of large-vessel vasculitis. The coexistence of isolated anti-Ro52 positivity and LVV in our patient may therefore indicate a shared interferon-driven inflammatory mechanism13.
The presence of persistent pulmonary consolidation necessitated careful consideration of infectious etiologies, particularly tuberculosis, given the patient’s impending long-term immunosuppression. Despite a negative GeneXpert test, a trial of anti-tuberculous treatment was initiated as a precautionary measure, reflecting the clinical dilemma in distinguishing between active vasculitis and infection in immunosuppressed or soon-to-be immunosuppressed patients. The dramatic clinical improvement and normalization of fever with intravenous methylprednisolone strongly supported the vasculitic nature of the symptoms.
Conclusion and Learning Points
This case illustrates a rare and complex presentation of Takayasu arteritis with concurrent Sjögren’s syndrome, initially masked by symptoms suggestive of infective endocarditis. Key learning points include: 1 The importance of a thorough physical examination, recognizing subtle signs like unequal pulses and blood pressure, and vascular bruits, which are critical for early suspicion of large-vessel vasculitis in patients presenting with AR and constitutional symptoms. 2 The necessity of timely advanced imaging, such as CT aortography, to confirm vascular involvement and exclude critical differential diagnoses like aortic dissection. 3 Takayasu arteritis may rarely coexist with Sjögren’s syndrome and should be considered even in the absence of classical sicca symptoms 4 Elevated inflammatory markers and pulmonary infiltrates require thorough exclusion of infection, particularly tuberculosis, before initiating or escalating immunosuppressive therapy. This case highlights the importance of careful clinical assessment and a multidisciplinary approach in achieving an accurate diagnosis and timely treatment of complex autoimmune vasculitis, thereby reducing the risk of long-term complications
References
- Bulut Gökten D, Yümün Kavak F, Soğur ÖA, et al. (2026) Coexistence of Sjögren’s disease and Takayasu arteritis in a woman: a case-based review. Clin Rheumatol. 45(1):123-129.
- Tshering P, Dorji T, Sangay. (2025) A case of isolated aortic regurgitation, later diagnosed as Takayasu arteritis: an importance of physical examinations. Clin Case Rep.;13(1):e71641.
- Verweij H, van der Veen MJ, van der Meer J. (2012) Late onset Takayasu arteritis and rheumatoid arthritis. Case Rep Rheumatol.;2012:523218.
- Naeem F, Imami SK, Khan SEA, et al. (2024) Atypical initial presentations of Sjogren’s syndrome: a case series. J Pak Med Assoc.;75(1):125-130.
- Unnikrishnan G, Hiremath N, Kesavadas C. (2018) Cerebral large-vessel vasculitis in Sjogren’s syndrome: utility of high-resolution magnetic resonance vessel wall imaging. J Clin Neurol.;14(4):588-591.
- Naeem FN, Saeed MA, Khan SEA. (2020) Coexistence of Takayasu arteritis with Sjogren’s syndrome initially presented with erythema nodosum. J Pak Med Assoc.;70(8):1477-1479.
- Jindal H, Suresh V, Kamaraj B, et al.(2025) Takayasu’s arteritis with systemic lupus erythematosus: a case report. Clin Med Insights Case Rep;19:11795468251350222.
- Andrzejewska K, Starba A, Misterska-Skóra M, et al. (2018) Palpable mass of the neck in the course of Takayasu arteritis. Reumatologia.;56(1):46-49.
- Kim WJ, Lee JS, Oh S. (2023) Progressive cerebral large-vessel vasculitis in a patient with Sjögren’s syndrome. J Neurocrit Care;16(1):60-63.
- Fath AR, et al. (2022) Surgical management of aortic regurgitation in Takayasu’s arteritis: a systematic review of techniques and outcomes. Tex Heart InstJ;49(5):e217017.
- Kaku Y, et al. (2015) Surgery for aortic regurgitation and aortic root dilatation in Takayasu arteritis. Asian Cardiovasc Thorac Ann;23(8):901-906.
- McGraw S, Tarter L, Farzaneh-Far A. (2015) Aortic regurgitation in Takayasu’s arteritis. QJM;108(5):421-422.
- Zhang H, et al. (2018) Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation;137(18):1934–1948. doi:10.1161/CIRCULATIONAHA.117.030423
Author Information
May Kyi Oo, Ei Pyaye Kyaw, Min Zaw Oo
- Senior Consultant physician, Medicine Department, Yangon General Hospital, Yangon
- Specialist Assistant Surgeon, Department of Medicine, Yangon General Hospital, Yangon
- Professor, Medicine Department , Yangon General Hospital, Yangon



